Applicant
Prof. Dr. Tilo Wettig
Universität Regensburg
Fakultät für Physik
93040 Regensburg
Project Summary
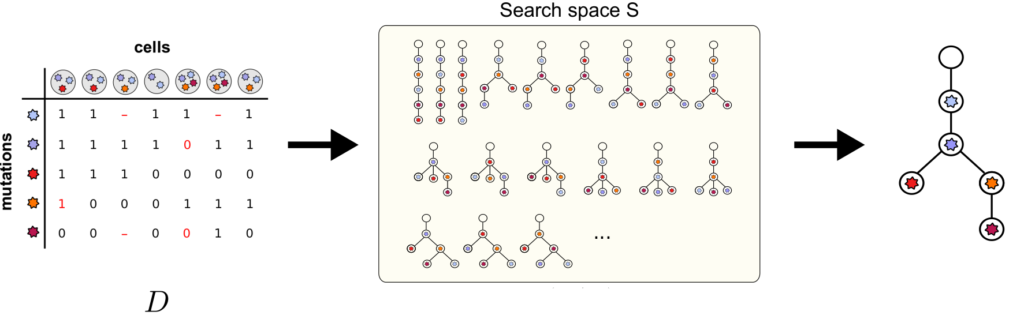
The recent emergence of high-throughput single-cell sequencing technologies has led to an unprecedented data explosion in the field of computational oncology. This development in combination with the high noise profiles inherent to single-cell measurements led to a number of computationally challenging problems. One such problem is the inference of mutation histories of tumours from high-throughput single-cell DNA sequencing data. A tumour develops through uncontrolled cell divisions that are first enabled and later exacerbated by a continuous acquisition of genetic alterations. Since these mutations happen at the level of individual cells and are passed on to descendent cells, this process creates a complex pattern of genetically distinct yet related cell populations referred to as clones and subclones. This genetic diversity poses a major challenge to targeted cancer therapies which often fail to eradicate the tumour in its entirety leaving minute resistant subclones that regrow the tumour and eventually lead to patient death. Knowing the mutational history and clonal composition of a tumour allows one in principle to adapt the therapy according to the clonal composition and improves the chances of a successful outcome. Due to the high noise level found in single-cell mutation calls, most methods employ a probabilistic search scheme to characterise not only the most likely mutational history of a tumour but also the uncertainty in the inference. Unfortunately for larger datasets comprising the mutational information of 10,000s of cells, the runtimes of these methods can become prohibitive. Here, we study a specific algorithm named SCITE and its successor ∞SCITE to show how high-performance computing can be used to enhance performance in the presence of large datasets. Our optimizations will be integrated in the SCITE/∞SCITE codebase.