

Applicant
Prof. Dr. Dirk Zahn
ComputerChemieCentrum – LS Theoretische Chemie
Friedrich-Alexander-Universität Erlangen-Nürnberg
Project Summary
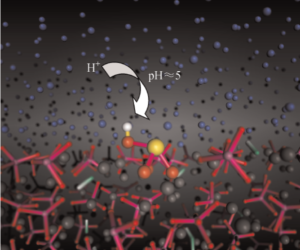
The achieved code implementation to LAMMPS (open source molecular dynamics simulation package) enables pH-dependent molecular simulations by means of parallel replica modeling. So far, the established parallel-replica scheme was based on different temperature and/or different interaction potentials, only. Now, different protonation states can be evaluated in parallel and be subjected to ‘local pK’ considerations. For the latter, we introduced an efficient quantum mechanical/molecular mechanical (QM/MM) assessment that combines the MM calculations of LAMMPS with prior QM characterization of small proxy models.
Key developments of the present Konwihr project are i) the creation of the parallel replica code and its handling of a master system in which protonation state is adapted to the desired pH of the system under investigation. Furthermore, ii) a series of sampling strategies for efficient assessment of the various replica has been developed. Future developments will aim at the more automated and simple-to-use implementation of our codes to ease the broader application of the pH-dependent simulation module.
The perspectives for the scientific community (applications range from chemistry, physics and biology to particle engineering and pharmaceutical formulation) are illustrated by successful grant proposals, both from public funds (DFG ZA 420-14/1 and ZA 420-16/1) and industry (Procter&Gamble study on the stabilization of calcium carbonate solutions in dishwasher application).